
语言模型在生物研究中的应用
Information
Language models for biological research: a primer |
||
Author:Elana Simon, Kyle Swanson & James Zou |
||
Jounal:Nature Methods |
Year:2024 |
|
Methods and Backgrounds
- Language Model: 语言模型可以学习序列中的复杂模式,例如句子中的单词和蛋白质中的氨基酸。由于这些模型大多是在异构序列集合上训练的,因此它们可以学习灵活的模式,并且可以适应性解决各种特定问题。由于其适应性,因此通常作为支持广泛下游应用程序的基础模型。其训练过程往往包括预训练和微调两个部分,其中对更大的数据集进行预训练(无监督)为模型提供了对数据的基本理解,从而能够在微调期间更有效地学习新目标。
Abstract
本文旨在介绍、回顾语言模型(基于Transformer架构)在生物学研究中的作用,其中预训练模型通过三种常见方法被用于研究:1.直接预测;2.embedding分析;3.迁移学习。
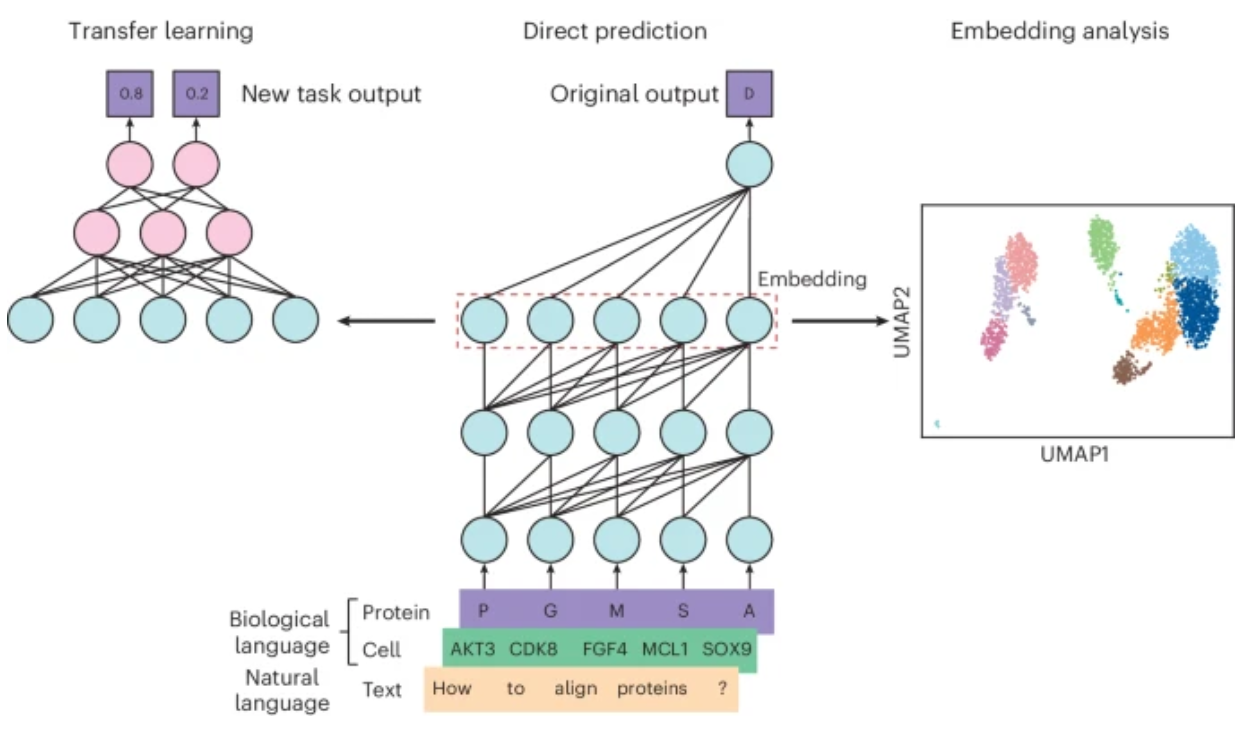 Fig. 1: Approaches to using language models for biological research.
Fig. 1: Approaches to using language models for biological research.
在本篇Primer中,作者介绍了如何使用这三种方法调整自然语言和生物语言模型来解决生物学中的一系列问题,以及使用语言模型进行生物学建模时的最佳实践,包括何时使用每种方法的建议以及给定当前模型限制需要考虑的注意事项。
Article Details
Natural Language Models
自然语言模型,例如ChatGPT,Claude之类的,在大量的数据集上训练,其中包括PubMed等生物来源。因此,它们可以微调为专业模型(例如,BioBERT或Med-PaLM2),为生物学研究者提供帮助。值得注意的是,因为ChatGPT、Claude这样的模型使用的训练数据集更广泛,因此其效果甚至可能优于Specialist的模型。
目前,自然语言模型可以大大加速生物学研究人员的效率,帮助阅读整理文献、总结文献中陌生概念,并可帮助完成生物信息学分析代码。
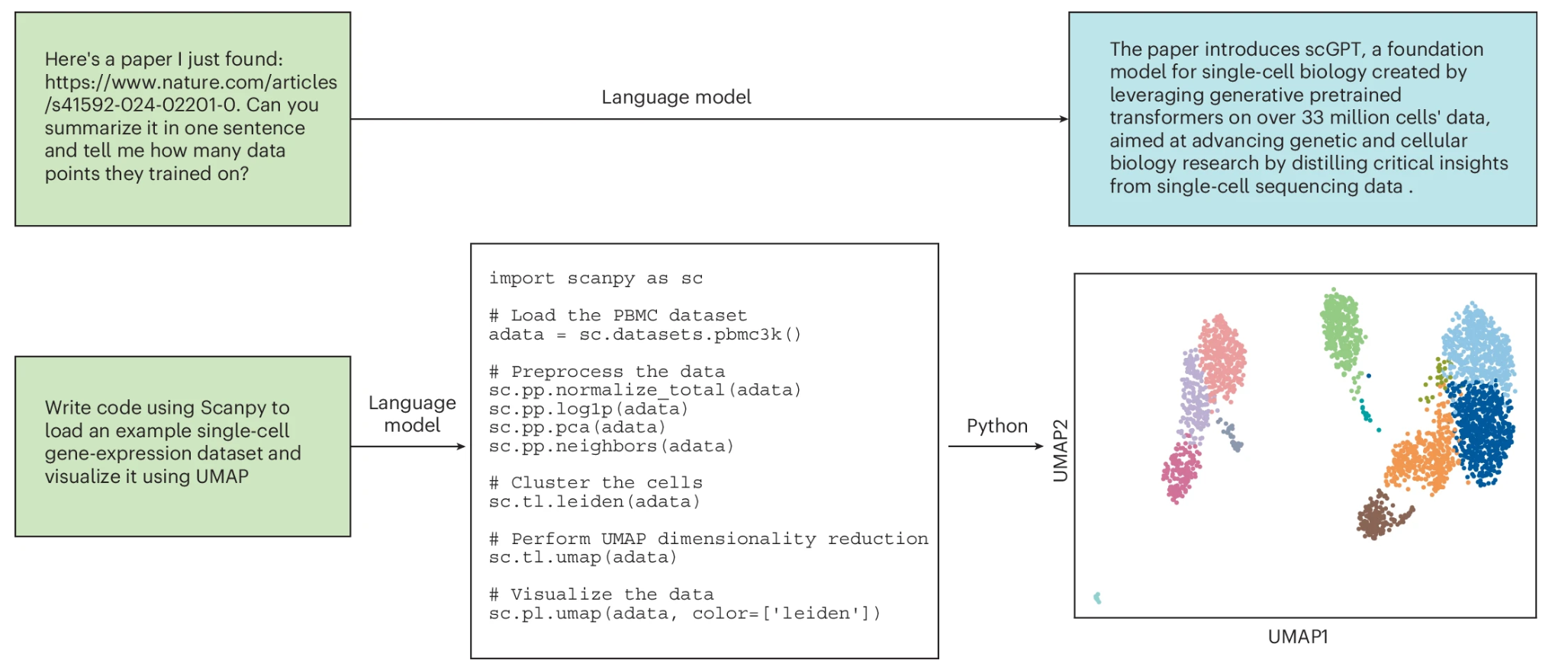 Fig. 2: Example uses of natural language models for biological research.
Fig. 2: Example uses of natural language models for biological research.
Biology Language Models
Protein Language Models
Importance of Large-scale pretraining
在蛋白质序列的大型数据集上预先训练的蛋白质语言模型可以学习捕获蛋白质进化约束和关键特性的表示。随后在较小的标记数据集上对这些模型进行微调,可以准确预测下游任务,例如稳定性、相互作用,甚至具有指定结构的序列设计。
Application
Direct Prediction:
根据这些模型的原始训练目标,可以使用这些模型直接预测每个氨基酸出现在序列中给定位置的概率。由于训练数据涵盖了已知功能性蛋白质序列的所有谱系,因此这些模型有效地学习了蛋白质进化的模式。如果不对突变的影响进行实验测量,该模型就可以隐式地了解哪些突变会对蛋白质功能有害,因为根据在整个进化过程中实证观察到的突变,它们被认为不太可能发生。因此,这些预测可以开箱即用地用于估计蛋白质编码突变的影响。
可以通过在给定位置特异性掩盖野生型氨基酸并要求模型在给定序列的其余部分来估算掩蔽位置来获得突变的可能性。如果根据语言模型,突变的可能性低于野生型氨基酸,则表明该突变可能是有害的。评估突变致病性的实验研究已经根据模型似然, 验证了这些估计。
将蛋白质序列建模为语言的一个好处是这些序列不需要事先比对或注释; 其他方法需要进化上对齐的蛋白质序列来预测突变的影响。蛋白质序列似然的语言模型估计也可用于估计蛋白质序列是否可能形成功能结构,这使得蛋白质语言模型能够评估和设计新的序列
Embedding Analysis
除了输出之外,蛋白质语言模型还提供有用的蛋白质嵌入。具体来说,当蛋白质序列通过模型运行时,可以提取模型对蛋白质中每个氨基酸的内部表示(嵌入)。然后,每个氨基酸的嵌入可以单独使用或组合成单个蛋白质表示。例如,先前的工作发现聚类蛋白质序列嵌入可以识别同源蛋白质。然后,可以根据每种蛋白质中单个氨基酸嵌入之间的相似性将这些同源蛋白构建成多序列比对。
Transfer Learning
由于微调蛋白质语言模型的成本很高,因此目前,更多的用法是将Embedding取出,输入给另一个更小的模型,以完成下游任务,例如:预测蛋白质稳定性、病毒抗原突变的免疫逃逸以及使用少量标记数据预测错义变体的致病性。或者,有更新、更高效的微调技术,使计算资源较少的研究人员能够完全微调大型蛋白质语言模型。
Protein Structure Models
虽然蛋白质结构预测模型,如 AlphaFold2和 ESMFold,不是本引物的重点,但值得一提的是,将结构信息与蛋白质序列一起包含在蛋白质序列中以训练模型,就像在蛋白质结构预测模型中所做的那样,可以改善各种下游任务的蛋白质表示。蛋白质结构预测模型与语言模型一样,已被证明可以通过直接预测、嵌入分析和迁移学习广泛适用于各种下游应用。
Single-cell Language Models
Importance of Large-scale pretraining
单细胞基因表达数据提供了对单个细胞的细胞状态和功能的见解,但它们的高维性使解释具有挑战性。最近开发了 AI 方法来帮助分析这些复杂数据。公开可用的单细胞基因表达数据的增长允许在不同细胞类型、组织、生物体和实验条件下的数百万个转录组上训练语言模型。这些模型可以泛化到新的数据集,并且可以针对各种下游任务进行微调,包括单元类型注释和批量校正。
Example: Geneformer
Geneformer就是这样一种单细胞语言模型。与许多其他生物语言模型一样,它具有经过训练的 transformer 架构,可为许多下游应用提供基因和细胞的表示。Geneformer 将每个细胞表示为细胞中表达量最大的 2,048 个基因的列表,根据 RNA 表达水平排序。训练过程与前面描述的蛋白质语言模型类似,因为基因的子集被掩盖,模型被训练以预测缺失的基因。为了按照表达水平的顺序正确预测缺失的基因,该模型必须了解各种基因的表达水平之间的相互作用,并隐式学习细胞类型特异性模式和背景。Geneformer 在跨越 40 种组织类型的 3000 万个单细胞转录组上进行了训练,这有助于它学习不同的表达模式。尽管 Geneformer 关注每个基因的相对表达水平,但其他单细胞语言模型使用替代公式。例如,单细胞语言模型 scGPT对定量表达值进行了预训练,从而实现了略有不同的下游应用。scGPT 还可以包括实验元数据,例如模态、批次和扰动条件。
Application
Direct Prediction:
单细胞语言模型的直接输出支持各种创造性的计算机实验。该模型可以通过获取单个细胞中按表达排序的原始基因列表、修改基因的顺序并量化这如何改变输出来估计遗传扰动对细胞的影响。例如,Geneformer 通过人为地将POU5F1、SOX2、KLF4 和 MYC 添加到细胞基因排名的顶部来模拟成纤维细胞的重编程,从而在计算上将细胞转移到诱导多能干细胞状态。同样,单细胞语言模型可以通过人为地从细胞的排名列表中删除基因并检查对细胞嵌入的影响来预测细胞对基因去除的敏感性。
Embedding Analysis
单细胞语言模型包含每个基因的嵌入,这些嵌入可以组合(例如,平均)以为每个细胞创建一个表示。这些细胞嵌入可用于聚类、可视化和细胞类型标记。由于训练数据的多样性和数量,这些模型可以在保持生物变异性的同时隐式减少批次效应,使它们能够从包含许多实验批次的数据集中识别细微的细胞亚型。
Transfer Learning
尽管这些嵌入中可能会出现有意义的簇,例如细胞类型,但也可以对模型进行微调以预测单个细胞的特性。例如,可以微调单细胞语言模型,以整合跨实验条件的数据并预测细胞类型标签和细胞状态。它们甚至可以支持基因的多模态表示。例如,可以对 scGPT 进行微调,以包括染色质可及性和蛋白质丰度以及基因表达水平,从而实现跨模态的数据集集成。
Multimodal Language Models for Biology
多模态模型可以跨多种数据模态(例如文本和图像)进行推理,从而使这些模型能够解决本质上涉及多种数据类型的任务。例如,病理学语言-图像预训练 (PLIP)在 Twitter 数据上进行了训练,以将病理图像与其字幕相匹配,使用户能够获取给定图像的字幕或查找给定文本描述的图像。同样,Med-PaLM Multimodal被训练为基于生物医学图像回答问题,而 MolT5被训练为根据分子结构用自然语言描述分子,包括有关其潜在生物学功能的信息。给定具有多种模态的数据点的足够示例,研究人员也可以为其他类型的生物数据训练多模态模型。
通过将生物文本的固定语言模型嵌入与来自其他领域的数据相结合,自然语言模型也可以应用于多模态设置,而无需额外训练。GenePT 为单细胞数据提供了一个示例。GenePT 利用语言模型的隐含基因组知识来嵌入细胞。具体来说,GenePT 使用 ChatGPT 嵌入细胞,首先使用 ChatGPT 嵌入来自 NCBI 的基因的文本描述,然后通过平均基于文本的基因嵌入来创建单细胞嵌入,按单细胞表达加权。在某些应用程序中,这些从自然语言模型派生的嵌入与来自生物语言模型(如 Geneformer)的嵌入相匹配或优于生物语言模型。类似的想法可以应用于生物学的其他领域;固定语言模型嵌入可以与来自替代模态的数据或模型合并,而无需额外训练。
Best practices when using language models for biology
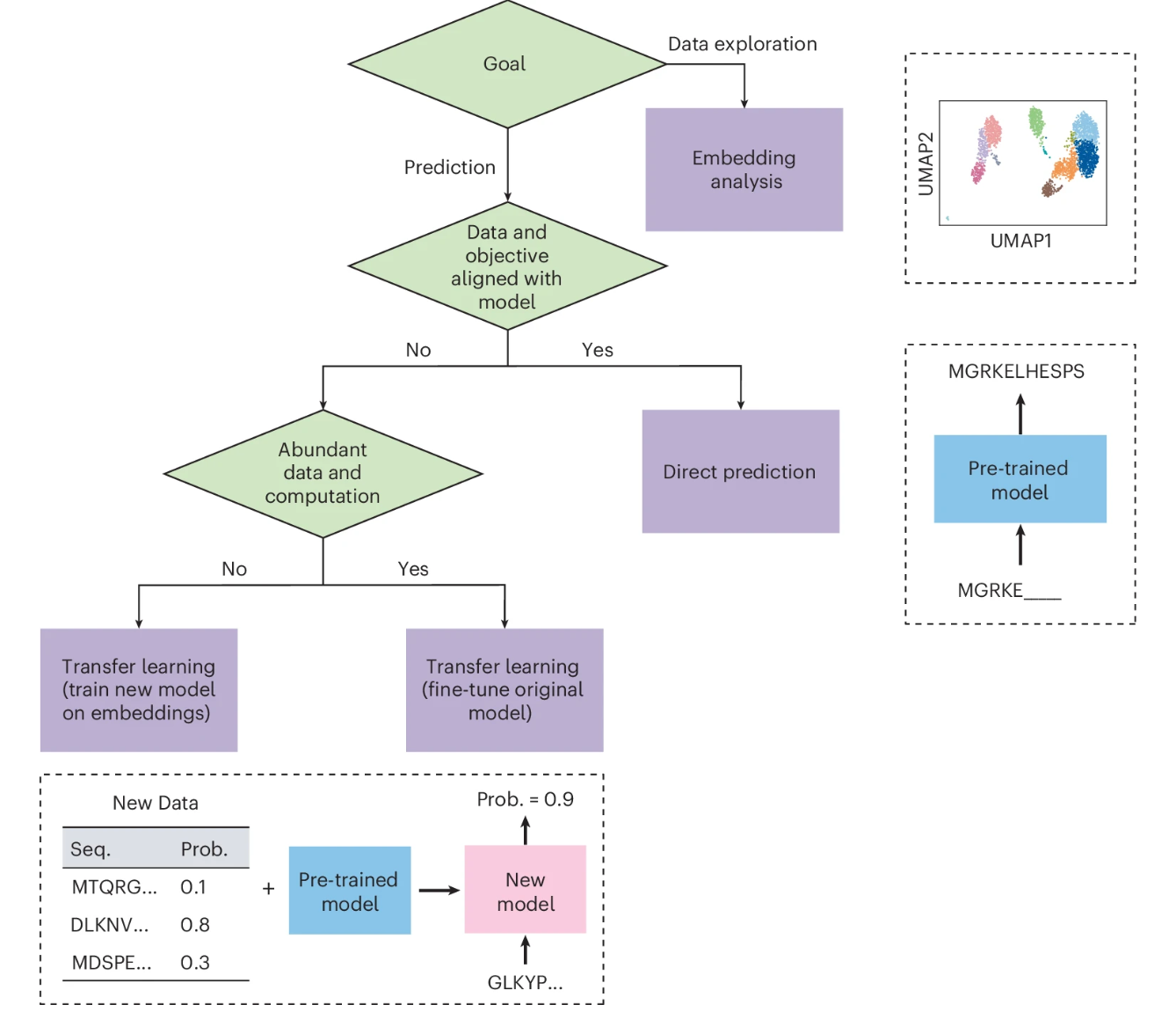 Fig. 3: Choosing the right approach for adapting a language model.
Fig. 3: Choosing the right approach for adapting a language model.
第一个决定是确定研究问题的目标。如果目标是数据探索,则可以使用嵌入分析方法以及降维和聚类等技术来揭示数据中的结构。但是,如果目标是根据数据进行预测,那么直接预测和迁移学习方法往往更有用。如果问题根据模型的训练数据和目标与模型的内在能力相匹配,那么直接预测方法是合适的,可能会根据目标修改输入。如果项目目标明显偏离模型的能力,或者如果有更特定于感兴趣任务的数据,那么迁移学习可能很有用。当有足够的数据和计算资源可用时,最好的方法可能是微调部分或全部语言模型。但是,如果数据或计算资源有限,另一种方法是使用语言模型计算新数据点的嵌入,并使用这些嵌入作为输入来训练单独的、通常较小的模型。此外,某些模型仅作为 Web 界面或应用程序编程接口 (API) 提供,这可能会限制其用于直接预测。其他具有开源代码和经过训练的模型参数的模型可用于嵌入分析或迁移学习。
一些模型还附带了 Jupyter 笔记本或 Google Colab 笔记本,演示了如何将预训练模型用于各种应用程序。该表罗列了它们中的部分。
尽管语言模型可能非常强大,但它们具有用户应注意的重要限制。首先,语言模型仍然无法完美地解决许多生物问题,即使是它们最初被训练要解决的问题。这可能是由于模型的限制,这些模型可能无法学习控制训练数据的所有模式,以及训练数据的限制。训练数据可能过时或有干扰,并且在某些类型的数据代表性不足的情况下,它们可能存在差距。例如,自然语言模型仅包含其训练数据中包含的生物知识,因此它们将不知道训练后发现的发现。蛋白质语言模型通常在标准氨基酸上进行训练,因此无法反映输入表示中任何翻译后修饰的重要性。单细胞表达数据可能很嘈杂,而资金优先级可能会使数据量偏向于特定的组织类型和疾病状态,这两者都是影响模型性能的因素。
此外,为特定生物应用量身定制的模型有时仍优于生物语言模型,尤其是当先验知识可以为模型设计提供信息时。例如,包含蛋白质结构信息的方法已被证明优于使用在蛋白质序列上训练的语言模型的方法。最后,评估适应其他任务的语言模型的性能也需要小心。语言模型是在大量可能不会公开共享的数据上进行训练的,因此确保语言模型的训练数据与下游任务的测试数据之间没有数据泄漏可能具有挑战性。



